A Enzimpótló kezelés Lizoszomális tároló betegségek kezelésére alkalmazzák, amelyekben az enzimek hiánya a bomlástermékek kóros felhalmozódásához vezet a sejtek lizoszómáiban.
A genetikai rendellenességek miatt hiányzó enzimeket rendszeres intravénás infúzióval kell kompenzálni. Mivel az infúzióban levő szintetikus enzimek molekuláris mérete miatt nem tudják átjutni a vér-agy gáton, a kezelés csak olyan lizoszomális tároló betegségek esetén működik, amelyek nem érintik a központi idegrendszert.
Mi az enzimpótló kezelés?

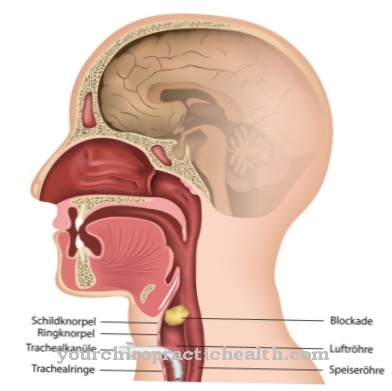
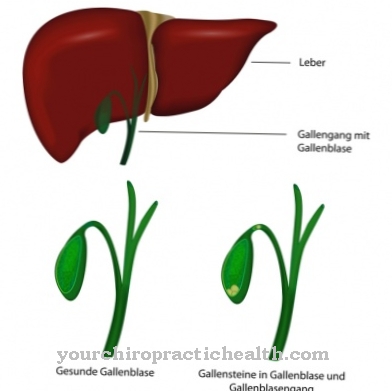
A lizoszómák olyan speciális sejt-organellák, amelyekben az idegen és endogén anyagok lebontásra és részleges újrahasznosításra kerülnek. Az anyagok lebontásához és szállításához speciális hidrolizáló enzimekre van szükség. Ezek proteázok, nukleázok, lipázok és transzporter anyagok.
Számos ismert genetikai hiba bizonyos enzimek kudarcához vezethet, így egyes bomlástermékek patológiás mennyiségben felhalmozódnak a lizoszómákban és felhalmozódnak, amíg ellenőrizetlenül el nem érik az extracelluláris mátrixot, azaz az intercelluláris tereket. Az összes genetikai hibát, amely legalább egy szükséges hidroláz meghibásodásához vezet, a lizoszomális tároló betegség kifejezés foglalja össze. Az enzimpótló kezelés (ERT, enzimpótló kezelés) a hiányzó endogén enzimek helyettesítésére szolgál szintetikusan előállított enzimekkel.
Mivel a hidrolázok viszonylag nagy molekulákból állnak, ezért nem képesek felszívódni a bélből anélkül, hogy először lebontnák és inaktiválnák, így csak intravénás infúzió útján adhatók be. Az enzimmolekulák mérete ugyanakkor megakadályozza a vér-agy gát átlépését, így a kezelés csak olyan lizoszomális tároló betegségek esetén lehet hatásos, amelyek nem érintik a központi idegrendszert (CNS).
Funkció, hatás és célok
Több mint 50 különféle lizoszomális anyagcsere-rendellenesség ismert, amelyek mindegyike monogenetikai hibára vezethető vissza. A lizoszomális tároló betegségeket hét különféle osztályra lehet osztani, a meglévő enzimhiány miatt a túlzottan tárolt anyagoktól függően.
A mukopoliszacharidózisok és az oligoszacharidózisok elsősorban az ERT-re alkalmasak. Az ERT célja az, hogy a specifikus enzimhiányt mesterségesen biztosított enzimekkel ellensúlyozza, hogy a betegség leálljon, vagy legalább enyhébb legyen. Részletesebben, helyettesítő enzimek állnak rendelkezésre a következő lizoszomális tároló betegségek esetén:
- Gaucher-kór
- Pompe-betegség
- Fabry betegség
- Hurler-Pfaundler szindróma (mucopolysaccharidosis I)
- Vadász-betegség (mucopolysaccharidosis II)
• Maroteaux-Lamy szindróma (mucopolysaccharidosis VI) • Niemann-Pick
Gaucher-kór a leggyakoribb lizoszomális tároló betegség. Három különböző változatban fordul elő, amelyek közül kettő az idegrendszert is befolyásolja. A nem neuropátiás formában különösen a lép lép be, amely nagymértékben megnő és másodlagos károsodásokhoz vezet, például vérszegénységhez és a csontvelő károsodásához. Jellemző tünetek a csonti és ízületi fájdalmak, valamint a keringési rendellenességek. A betegség akut neuropátiás változata súlyos lefolyású, és kevés esélyt kínál a túlélésre az élet első két évében.
A Pompe-betegség a betegség oka az alfa-1,4-glükozidáz enzim hiánya, amely számos metabolikus folyamatban részt vesz. A Pompe-betegség a szív hatalmas megnagyobbodásához (kardiomegalia) és a szívelégtelenséghez vezet. Vannak korai, komoly tanfolyamok, amelyek az élet első néhány hónapjában jelennek meg, valamint enyhébb formák, amelyek csak az élet későbbi éveiben jelentkeznek.
A Fabry-betegséget egy X-hez kapcsolódó genetikai hiba okozza, így a tárolási betegség csak fiúkat és férfiakat érinti. A betegség általában előrehaladott gyermekkori tünetekhez vezet, ideértve a fájdalomtámadásokat, a bőr keratómáit, veseproblémákat és a szívizomkárosodásokat. Az alfa-galaktozidáz A enzim hiánya a ceramid-trihexozid felhalmozódásához vezet, amely a tünetek kiváltásának oka, amely befolyásolhatja az autonóm idegrendszert is.
Nem ritka, hogy a kár szívrohamhoz, veseinfarktushoz vagy akár stroke-hoz is vezethet. A Hurler-Pfaundler szindrómát I típusú mukopoliszacharidózisnak is nevezik, és a glikozaminoglikán metabolizmusának megszakadása okozza. A betegség számos tünettel társul, beleértve a súlyos mentális károsodást és a súlyos csontváz-változásokat. A betegség lefolyása súlyos, így az átlagos várható élettartamot 11–14 évnek tekintik. A Hunter-betegség a 2. típusú mucopolysaccharidosisnak felel meg, és - akárcsak Hurler-kór - egy X-hez kapcsolódó hibának okozta. A betegséget különböző súlyosságú folyamatok jellemzik, kezdve a korai gyermekkorban és az enyhe kimenetelekkel, amelyek csak felnőtt férfiakon jelentkeznek.
A leggyakoribb szív tünetek, mint például a szívbillentyű-rendellenességek és a szívizomproblémák miatt, a várható élettartam a normálistól kissé korlátozottig terjed. A Maroteaux-Lamy szindróma (MPS VI) egyike azoknak a mukopoliszacharidózisoknak, amelyeket autoszomális recesszív módon örökölnek, mivel az okozati génhiány nincs az X kromoszómán. A betegség nagyon ritka, 455 000 születésenként egy eset fordul elő. Ismertek enyhe és súlyos formák.
A tünetek a megnagyobbodott máj és lép, a carpalis alagút szindróma és a szívbillentyűk változásai. A Niemann-Pick B szfingomyelin lipidózis, amely az egyik lizoszomális tároló betegség, és amelyet a 11. kromoszóma genetikai hibája okoz. Míg a B-típusú betegség elsősorban a májat és a lépt érinti, az A-típusnak jelentős idegproblémái is vannak.
Itt megtalálja gyógyszereit
Pain fájdalomcsillapítókKockázatok, mellékhatások és veszélyek
Mivel az enzimpótló terápiával kezelhető lizoszomális tároló betegségek közül sok súlyos folyamatot vesz igénybe, ennek megfelelően megnövekedett halálozási arány mellett, az ERT-ben a legnagyobb kockázat az, hogy a kiválasztott pótló enzim nem működik, vagy csak túl gyengén működik.
Egy másik kockázat kevésbé magában a terápiában rejlik, mint abban a tényben, hogy az alapbetegséget túl későn ismeri fel, így az ERT megállhat a kurzus folyamán, de a már okozott kár nem állhat vissza. Körülbelül minden második kezelt beteg ideiglenesen reagál az infúzióra olyan tünetekkel, mint például láz és hidegrázás. Ennek okai még nem tisztázottak teljesen. Egyes betegek antitestek kialakulásával reagálnak, és ismertek olyan esetek, amikor a betegek kiütésekkel és hörgőgörcsökkel reagáltak.